酶促反应
酶促反应(又称酶催化)是指由一类被称为酶的特殊蛋白质所催化的化学反应。因为非催化反应的速率特别慢,故细胞中生物化学反应的催化作用就显得极重要。
酶促反应的机制与其他类型的化学催化在原理上很相似。酶通过提供替代反应路线以及稳定中间产物的方法,减少了为达到最高能量过渡态时的能量需求。活化能(Ea)的减少增加了具有足够达到活化能并形成产物的反应物分子的数量。
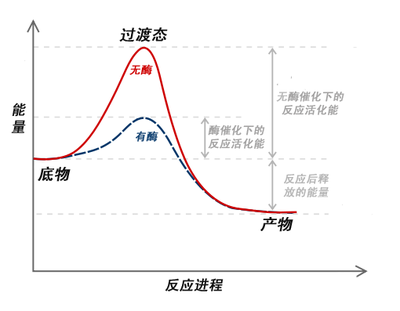
酶使得中间状态达到稳定。
目录
1 诱导契合
1.1 诱导契合催化
2 过渡态的稳定机制
2.1 键扭曲催化
2.2 临近效应与定向效应
2.3 涉及质子供体与受体的催化(酸碱催化)
2.4 静电催化
2.5 共价催化
2.6 量子隧穿效应
3 催化机制的例子
3.1 磷酸丙糖异构酶
3.2 胰蛋白酶
3.3 醛缩酶
4 参考文献
5 延伸閱讀
6 参见
7 外部連結
诱导契合

酶反应诱导契合学说的图解。
最受欢迎的酶-底物反应模型是诱导契合模型[1]。该模型提出了:酶和底物之间的最初反应是相对较弱的,但这些弱反应迅速引导酶中的构象改变以使结合变得更加紧密。
诱导契合催化
因为酶紧密结合的稳定效应,诱导契合机制的优势就体现了出来。底物结合有两种不同的机制:①、一致结合,酶与底物结合很紧密;②、差别结合,酶与过渡态结合很紧密。一致结合的稳定结果是同时增加了底物和过渡态的结合亲和力,然而差别结合只增加过渡态的结合亲和力。这两者都在进化中被酶所采用并用以将反应活化能降到最低。被饱和状态的酶,即与底物有高度结合亲和力的状态时就需要差别结合以减少活化能,然而未与酶结合的小底物可采用一致结合或是差别结合两者中的一种。
这些效应引导大多数蛋白质采用差别结合机制以减少活化能,因此大多数蛋白质具有酶与过渡态的高度亲和力。差别结合是由诱导契合机制所实行起来的——底物首先与之相弱结合,然后酶改变其构象,增加了与过渡态的亲和力并稳定这个过渡态,因此减少了达到过渡态所需的活化能。
需要澄清的是:诱导契合的概念并不能对催化加以科学的说明。即,化学催化被定义为在水中(没有酶)非酶促情况下Ea‡(当系统处于ES‡的状态)相对于Ea‡发生降低。而诱导契合仅意味着在结合酶形态下壁垒会降低,但并没有告诉我们使得反应壁垒降低的原因是什么。
诱导契合可能有利于解释:当竞争和干扰存在时可通过构象校对机制保证分子识别的精确性[2]。
过渡态的稳定机制
这些构象改变也可以用于使活性位点中的催化残基靠近那些在反应中会被改变的底物中的化学键。在结合发生后,通过提供反应替代化学路径的方法,一种或多种催化机制使得反应过渡态的能量降低。有六种可能的“越过壁垒”或“通过壁垒”的催化机制:
键扭曲催化
这种作用对诱导契合结合有着主要影响,在该情况下酶对过渡态的结合力比对底物本身的结合力要强。这种作用诱导了结构重排,即将底物的键扭曲成一种接近过渡态的构象,因此降低了底物与过渡态之间的能量差距以帮助催化此反应。
然而,事实上,称这种扭曲为“基态失稳效应”比“过渡态稳定效应”更贴切[3][4]。此外,酶是非常灵活的并且自己不会有较大的形变效应[5]。
除了底物中的键扭曲外,键扭曲同时可以被酶本身所采用以激活活性位点的残基。
例如: |
溶菌酶的底物、结合底物与过渡态构象 |
底物在与酶结合时由典型的己糖环“椅式构象”被扭曲为“沙发式构象”,在形状上与过渡态更相似了。 |
临近效应与定向效应
这个效应以使酶-底物相互作用的反应化学基团对齐并将它们靠在一起的方式加快了反应速率。这减少了反应物的熵,这使得连接反应或加成反应反应地更顺利,当两个反应物变为单个产物时,全部熵损失就会减少。
这个效应可以类比为增加了反应物浓度的效应。这种底物与反应物的结合赋予了这种反应分子内进行的特性,激增了反应速率。
例如: |
如果相似的反应是在分子内进行的,那么反应会大大加快 |
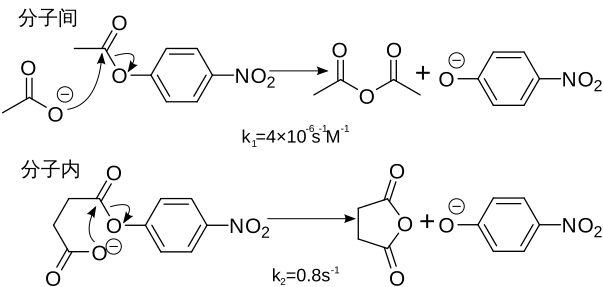 |
在分子内反应中,乙酸盐的有效浓度估计可以达到k2/k1 = 2×105摩尔。 |
然而,这种情况可能会变得更加复杂,因为现代计算研究已公认传统的临近效应例子并不与酶的熵效应直接挂钩[6][7][8]。并且,已发现最初的熵理论[9]对催化定向效应熵的贡献是被大大高估了的[10]。
涉及质子供体与受体的催化(酸碱催化)
质子供体与受体,如酸或碱,可以供出或接受质子,以使过渡态发展中电荷达到稳定。这是典型的激活亲核与亲电基团或是稳定离开基团的效应。组氨酸是常见的涉及到酸/碱反应的残基,因为其酸度系数接近于中性且因此可以既可作为质子的受体又可作为质子的供体。
许多涉及到酸/碱催化的反应机制会采取基本上变动的酸度系数。这个酸度系数的变动可以通过残基的局部微环境来实现。
| 环境状态 | 酸 | 碱 |
|---|---|---|
| 疏水环境 | 增加酸度系数 | 减少酸度系数 |
| 临近相似电荷的残基 | 增加酸度系数 | 减少酸度系数 |
| 盐键(与疏水键)构造 | 减少酸度系数 | 增加酸度系数 |
酸度系数可被环境明显地改变,在一定程度上在溶液中是碱性的残基也可能作为质子供体,并且反之亦然。
例如: |
丝氨酸蛋白酶催化机制 |
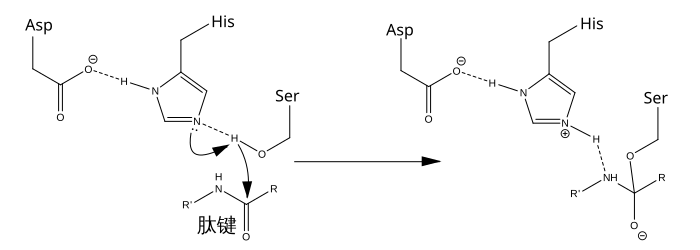 |
丝氨酸蛋白酶催化机制的最初步骤涉及到活性位点的组氨酸从丝氨酸残基上接受一个质子。这一步骤将丝氨酸装备成一个亲核试剂以备攻击底物中的酰胺键。这一机制包括了自丝氨酸(一种碱,酸度系数为14)供出的质子转移到组氨酸(一种酸,酸度系数为6)上,因为微环境的碱性使得此过程得以发生。 |
有必要澄清的是:酸度系数的改变完全是静电机制的一部分[4]。此外,上述例子中的催化效应主要与氧阴离子酸度系数的减少以及组氨酸酸度系数的增加有关,而质子自丝氨酸转移到组氨酸上的作用并不是显著的催化作用,因为它不是决定屏障的速率[11]。
静电催化
带电过渡态的稳定亦可以通过使活性位点中的残基与中间产物之间形成离子键(或局部离子电荷相互作用)而达到。这些键来自于例如赖氨酸、精氨酸、天冬氨酸或谷氨酸等氨基酸上面的酸或碱侧链,或是来自于如锌一类的金属辅因子。金属离子是特别有效的,并且可以将水的酸度系数减到足够低以使其变为一个有效的亲核试剂。
系统计算机模拟研究确定了静电效应到目前为止是对催化作用贡献最大的效应[4]。尤其是实践已证明酶可以提供比水极性更大的环境,并且离子态过渡态是被固定偶极子所稳定的。这与水中过渡态的稳定情况很不相同,在后者的情况下水分子必须付出“重组能”[12],才得以使离子态和过渡态达到稳定。因此,催化作用是与酶预组织起来的极性基团相关的[13]。
例如: |
羧肽酶催化机制 |
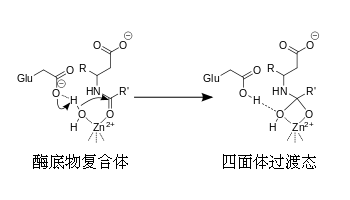 |
四面体中间产物被是被锌离子与氧原子上的负电荷之间的离子键所稳定起来的。 |
共价催化
共价催化涉及到底物与活性位点残基或是辅酶之间形成一个短暂存在的共价键。这使得在反应中多了一个额外的共价中间体,这有助于降低后面反应过渡态的能量。在反应之后的阶段,共价键必须必须被打断以使酶再生。在如胰凝乳蛋白酶和胰蛋白酶等的蛋白酶中发现了这一机制,在反应中形成了酰基酶中间产物。使用来自赖氨酸残基的自由胺形成施夫碱的过程则是另外一种机制,就像在糖酵解过程中的醛缩酶一样。
一些酶利用如磷酸吡哆醛(PLP)或硫胺素焦磷酸(TPP)之类的非氨基酸辅因子与反应物分子间反应形成共价中间产物[14][15]。这样为降低后面反应过渡态能量而形成的共价中间产物在功能上与反应物与活性位点中的氨基酸残基形成共价中间产物是一样的,都是为了达到稳定;而所不同的是辅因子有着允许带着产物离开酶之功能然而氨基酸侧链则不行。酶利用这样辅因子的例子包括:依赖磷酸吡哆醛的酶天冬氨酸转氨酶和依赖硫胺素焦磷酸的酶丙酮酸脱氢酶[16][17]。
有必要澄清的是:共价催化有时仅相当于是利用一种特殊的机制,而不是真正的催化[4]。例如,胰凝乳蛋白酶中连接到活泼丝氨酸分子上的共价键应被类比为已被充分理解的非催化溶液反应中连到亲核试剂上的共价键。真正共价催化(在这种情况下能量壁垒比在相应非酶促溶液中的要低)理论是需要,例如,一个由酶基团连接到过渡态的局部共价键(例如一个非常强的氢键),并且这种效应对催化来说并贡献并不明显。
量子隧穿效应
由于“通过壁垒”机制(量子隧穿效应)模型和观察资料的出现,这些传统的“越过壁垒”机制在某些情况下受到挑战。一些酶运转地比经典的ΔG‡所预测的动力学参数还要快。在“通过壁垒”模型中,一个质子或电子可以隧穿过活化能壁垒[18][19]。质子的量子隧穿效应已经在氧化色胺的芳香胺脱氢酶中被观察到[20]。
有趣的是,量子隧穿效应似乎没有对酶促反应有很大的帮助,因为隧穿效应对酶促反应与溶液中的非酶促反应同时做着贡献[19][21][22][23]。然而,隧穿贡献(通常相较于经典的“越过壁垒”途径的反应速率来说增加为原来的1000倍[24])对于生物有机体生存能力来说可能是关键因素。这强调了隧穿作用对生物的特别重要性。
在1971年-1972年间,首个酶促反应的量子力学模型被系统地阐述出来[25][26]。
催化机制的例子
在实践中,大多数酶机理涉及到不同类型催化相结合在一起。
磷酸丙糖异构酶
磷酸丙糖异构酶(EC 5.3.1.1)催化两种丙糖磷酸异构体——磷酸二羟丙酮与D-3-磷酸甘油醛之间的可逆转换。
胰蛋白酶
胰蛋白酶(EC 3.4.21.4)是一种可以断开蛋白质底物赖氨酸与精氨酸氨基酸残基之间键的丝氨酸蛋白酶。
醛缩酶
醛缩酶(EC 4.1.2.13)催化果糖-1,6-二磷酸分解成甘油醛-3-磷酸与磷酸二羟丙酮。
参考文献
^ Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis.. Proc. Natl. Acad. Sci. U.S.A. February 1958, 44 (2): 98–104. PMC 335371. PMID 16590179. doi:10.1073/pnas.44.2.98.
^ Savir Y & Tlusty T. Conformational proofreading: the impact of conformational changes on the specificity of molecular recognition (PDF). PLoS ONE. 2007, 2 (5): e468. PMC 1868595. PMID 17520027. doi:10.1371/journal.pone.0000468.
^ Jencks W.P. "Catalysis in Chemistry and Enzymology" 1987, Dover, New York
^ 4.04.14.24.3 Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H.M. Electrostatic Basis of Enzyme Catalysis. Chem. Rev. 2006, 106 (8): 3210–3235. PMID 16895325. doi:10.1021/cr0503106. 引用错误:带有name属性“warshel”的<ref>标签用不同内容定义了多次
^ Warshel, A.; Levitt, M. Theoretical Studies of Enzymatic Reactions: Dielectric Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme. J. Mol. Biol.: 227.
^ Stanton, R.V.; Perakyla, M.; Bakowies, D.; Kollman, P.A. Combined ab initio and Free Energy Calculations To Study Reactions in Enzymes and Solution: Amide Hydrolysis in Trypsin and Aqueous Solution. J. Am. Chem. Soc. 1998, 120: 3448–3457. doi:10.1021/ja972723x.
^ Kuhn, B.; Kollman, P.A. QM-FE and Molecular Dynamics Calculations on Catechol O-Methyltransferase: Free Energy of Activation in the Enzyme and in Aqueous Solution and Regioselectivity of the Enzyme-Catalyzed Reaction. J. Am. Chem. Soc. 2000, 122: 2586–2596. doi:10.1021/ja992218v.
^ Bruice, T.C.; Lightstone, F.C. Ground State and Transition State Contributions to the Rates of Intramolecular and Enzymatic Reactions. Acc. Chem. Res. 1999, 32: 127–136. doi:10.1021/ar960131y.
^ Page, M.I.; Jencks, W.P. Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect. Proc. Natl. Acad. Sci. USA: 1678–1683.
^ Warshel, A.; Parson, W.W. Dynamics of Biochemical and Biophysical Reactions: Insight from Computer Simulations. Quart. Rev. Biophys. 2001, 34: 563–679.
^ Warshel, A.; Naray-Szabo, G.; Sussman, F.; Hwang, J.-K. How do Serine Proteases Really Work?. Biochemistry: 3629.
^ Marcus R. A. "On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions" J. Chem. Phys. 1965 43:679-701
^ Warshel A. "Energetics of Enzyme Catalysis", Proc. Natl. Acad. Sci. USA, 1978 75: 5250
^ Toney, M. D. "Reaction specificity in pyridoxal enzymes." Archives of biochemistry and biophysics (2005) 433: 279-287
^ Micronutrient Information Center, Oregon State University
^ Voet, Donald; Judith Voet. Biochemistry. John Wiley & Sons Inc. 2004: 986–989. ISBN 0-471-25090-2. 引文使用过时参数coauthors (帮助)
^ Voet, Donald; Judith Voet. Biochemistry. John Wiley & Sons Inc. 2004: 604–606. ISBN 0-471-25090-2. 引文使用过时参数coauthors (帮助)
^ Garcia-Viloca, M; Gao, J; Karplus, M; Truhlar, DG. How enzymes work: analysis by modern rate theory and computer simulations.. Science. 2004, 303 (5655): 186–95. PMID 14716003. doi:10.1126/science.1088172.
^ 19.019.1 Olsson, MH; Siegbahn, PE; Warshel, A. Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase.. Journal of the American Chemical Society. 2004, 126 (9): 2820–8. PMID 14995199. doi:10.1021/ja037233l. 引用错误:带有name属性“Mats”的<ref>标签用不同内容定义了多次
^ Masgrau, L; Roujeinikova, A; Johannissen, LO; Hothi, P; Basran, J; Ranaghan, KE; Mulholland, AJ; Sutcliffe, MJ; Scrutton, NS. Atomic description of an enzyme reaction dominated by proton tunneling.. Science. 2006, 312 (5771): 237–41. PMID 16614214. doi:10.1126/science.1126002.
^ Hwang, J.-K.; Warshel, A. How important are quantum mechanical nuclear motions in enzyme catalysis. J. Am. Chem. Soc: 11745–11751.
^ Ball, P. Enzymes: By chance, or by design?. Nature: 396.
^ Olsson, M.H.M.; Parson, W.W.; Warshel, A. Dynamical Contributions to Enzyme Catalysis: Critical Tests of A Popular Hypothesis. Chem. Rev.: 1737–1756.
^ Masgrau, L; Roujeinikova, A; Johannissen, LO; Hothi, P; Basran, J; Ranaghan, KE; Mulholland, AJ; Sutcliffe, MJ; Scrutton, NS. Atomic description of an enzyme reaction dominated by proton tunneling.. Science. 2006, 312 (5771): 237–41. PMID 16614214. doi:10.1126/science.1126002.
^ Volkenshtein M.V., Dogonadze R.R., Madumarov A.K., Urushadze Z.D., Kharkats Yu.I. Theory of Enzyme Catalysis.- Molekuliarnaya Biologia, Moscow, 6, 1972, 431-439
^ Volkenshtein M.V., Dogonadze R.R., Madumarov A.K., Urushadze Z.D., Kharkats Yu.I. Electronic and Conformational Interactions in Enzyme Catalysis. In: E.L. Andronikashvili (Ed.), Konformatsionnie Izmenenia Biopolimerov v Rastvorakh, Publishing House "Nauka", Moscow, 1973, 153-157
延伸閱讀
.mw-parser-output .refbeginfont-size:90%;margin-bottom:0.5em.mw-parser-output .refbegin-hanging-indents>ullist-style-type:none;margin-left:0.mw-parser-output .refbegin-hanging-indents>ul>li,.mw-parser-output .refbegin-hanging-indents>dl>ddmargin-left:0;padding-left:3.2em;text-indent:-3.2em;list-style:none.mw-parser-output .refbegin-100font-size:100%
- Alan Fersht, Structure and Mechanism in Protein Science : A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, 1998. ISBN 0-7167-3268-8
Dedicated issue of Philosophical Transactions B on Quantum catalysis in enzymes freely available.[永久失效連結]
参见
- 酶活性测定
- 酶动力学
- 蛋白质动力学
- 量子隧穿效应
- 蛋白质水解地图
- 时间分辨晶体学
外部連結
 維基共享資源中与酶促反应相關的分類
維基共享資源中与酶促反应相關的分類
| ||||||||||||||||||
| ||||||||||||||||||||||


